 | |||||||||||||||
 |
 |
 |
|
 |
|
 |
|
 |
|
 |
|
 |
|
 |
 |
AMBER Archive (2009)
Subject: [AMBER] Can we use Amber?
From: Casey,Richard (Richard.Casey_at_ColoState.EDU)
Date: Thu Jan 08 2009 - 11:16:50 CST
- Next message: wl2290_at_columbia.edu: "[AMBER] mairun"
- Previous message: michael bane: "Re: [AMBER] how many processors for parallel test"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
Hello,
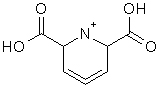
We would like to run molecular dynamics simulations for the compound shown in the attached file. Is Amber the correct force field for this? Or are there other more appropriate force fields?
--------------------------------------------
Richard
_______________________________________________
AMBER mailing list
AMBER_at_ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber
- Next message: wl2290_at_columbia.edu: "[AMBER] mairun"
- Previous message: michael bane: "Re: [AMBER] how many processors for parallel test"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ]
|
VU Home |
VUMC Home |
People Finder |
University Calendar
webmaster- modified on January 30, 2009 |